
Introduktionen av biosimilarer inom reumatologin
En biosimilar eller ”Biosimilar biological medicinal product” som är den officiella engelska beteckningen är ett läkemedel som liknar ett redan godkänt biologiskt läkemedel, det så kallade referensläkemedlet. Men en biosimilar är inte identisk i motsats till generika. Vad innebär detta i praktiken?
Den Europeiska läkemedelsmyndigheten (EMA) publicerade 2020 en uppdaterad vägledning för vårdpersonal som även är tillgänglig på svenska [1]. Den grundläggande informationen nedan är hämtad ur denna skrift. I denna vägledning beskriver man att EU har banat väg för lagstiftningen rörande biosimilarer sedan godkännandet av den första biosimilaren 2006.
När det gäller switch mellan olika biosimilarer har Läkemedelsverket precis publicerat en genomgång [2]. EMA konkluderar att erfarenheten från över 10 års klinisk erfarenhet visar att biosimilarer som godkänts av EMA kan användas lika säkert och effektivt som andra biologiska läkemedel för alla indikationer de godkänts för.
Flera biosimilarer har blivit tillgängliga för reumatologiska indikationer: de monoklonala antikropparna (infliximab, adalimumab och rituximab) samt fusionsproteinet etanercept. De första biosimilarerna som blev tillgängliga var för infliximab och etanercept. Därefter följde rituximab och adalimumab. Utöver dessa preparat är flera andra biosimilarer godkända inom EU (02/2019): lågmolekylärt heparin (enoxaparinnatrium), tillväxtfaktorer (epoetin, filgastrim och pegfilgastrim), hormoner (follitropin alfa, insulin glargin, insulin lispro, somatropin, teriparatid och även monoklonala antikroppar som används inom onkologin (bevacizumab och trastuzumab).
Molekylstorleken varierar dock påtagligt mellan dessa preparat. Insulin har en molekylstorlek på 5 808 dalton, tillväxthormon (somatropin) på 22 000 dalton och de monoklonala antikropparna 150 000 dalton. I regel är tillverkningen av större molekyler mera komplicerad och sannolikheten för viss variation, till exempel avseende glykosyleringsmönster, ökar. Detta är dock inte unikt för biosimilarer utan det gäller även referensprodukten. I och med att det kan förekomma viss variation har man dragit slutsatsen att en biosimilar ska betraktas som ett unikt läkemedel till skillnad från generika där den aktiva substansen är identisk med referenspreparatet.
För att förstå konceptet med biosimilarer är det bra att känna till att det under livscykeln för biologiska läkemedel sker förändringar av tillverkningsprocessen. Olika batcher av samma biologiska läkemedel kan uppvisa en liten variationsgrad till exempel när det gäller glykosylering. Aminosyrasekvensen däremot och proteinets aktivitet är densamma i alla batcher. För att granska dessa förändringar finns ett särskilt regelverk (Comparability excercise), som bland annat innebär att företagen ska visa att ändringar i tillverkningsprocessen inte har påverkat kvaliteten, effekten eller säkerheten negativt. Ur regulatorisk synvinkel skulle en biosimilar kunna betraktas som ytterligare en version av originalprodukten eftersom samma vetenskapliga principer tillämpas.
Vilka jämförbarhetsstudier krävs det efter ändringar i tillverkningsprocessen av ett läkemedel som framställs genom bioteknologi? En mindre ändring (till exempel introduktion av en känsligare testmetod för att analysera den aktiva substansen, som inte påverkar läkemedlets farmaceutiska kvalitet förutsätter begränsade fysikaliskt-kemiska studier som jämför batcher före och efter förändringen. En betydande ändring (till exempel förändringar i det cellsystem som används för att tillverka den aktiva substansen), som kan påverka läkemedlets egenskaper eller specifikationer men inte förväntas påverka säkerhet och effekt, förutsätter omfattande fysikalisk-kemiska och funktionella (in vitro) jämförbarhetsstudier. Större ändringar (till exempel vissa ändringar i läkemedlets sammansättning) som eventuellt kan påverka säkerhet och effekt förutsätter omfattande fysikalisk-kemiska studier och in vitro-studier som vid behov kompletteras med prekliniska och kliniska studier.
Vad krävs för att en biosimilar ska godkännas för försäljning? Biosimilaren och det biologiska referensläkemedlet får inte skilja sig åt i fråga om säkerhet och effekt vid behandling av patienter. En biosimilar måste alltså karaktäriseras såväl biokemiskt, prekliniskt och kliniskt.
Vilka egenskaper utmärker biosimilarer?
Likvärdighet med referensläkemedlet: Biosimilaren har fysikaliska, kemiska och biologiska egenskaper som är jämförbara med referensläkemedlets. Små skillnader, som inte är betydelsefulla vad gäller läkemedlets säkerhet och effekt, får förekomma.
Inga kliniskt betydelsefulla skillnader i förhållande till referensläkemedlet: Kliniska studier som ligger till grund för godkännandet av en biosimilar bekräftar att eventuella skillnader inte påverkar läkemedlets säkerhet och effekt.
Naturliga variationer i biosimilaren hålls inom strikta gränser: Små variationer är endast tillåtna om det finns vetenskapliga belägg som visar att de inte påverkar biosimilarens säkerhet och effekt. Den tillåtna variationen för en biosimilar är densamma som mellan olika batcher av referensläkemedlet. En robust tillverkningsprocess säkerställer att alla batcher av läkemedlet håller bevisad hög kvalitet.
Samma strikta normer för kvalitet, säkerhet och effekt: Biosimilarer godkänns enligt samma strikta normer för kvalitet, säkerhet och effekt som gäller för andra läkemedel.
Till skillnad från generiska läkemedel som godkänns på basen av farmakokinetiska bioekvivalensstudier utan behov av ytterligare kliniska data krävs det för biosimilarer såväl farmakokinetiska som farmakodynamiska studier, samt uppgifter om säkerhet och effekt. Effekt och säkerhet måste motiveras för varje indikation. Kliniska studier med biosimilaren är dock vanligen inte nödvändiga för varje indikation som har godkänts för referensläkemedlet. Efter att biosimilaritet har påvisats är extrapolering till andra indikationer möjlig om de vetenskapliga bevis som finns tillgodoser alla specifika aspekter hos indikationerna.
På vilket sätt skiljer sig kraven på biologiska läkemedel (till exempel referensläkemedel) från biosimilarer? För nya biologiska läkemedel krävs fullständiga prekliniska data avseende farmakologi och toxikologi samt konventionella kliniska prövningar avseende säkerhet och effekt för alla indikationer. För biosimilarer avgörs behovet av prekliniska data på resultaten av kvalitetsstudier. Jämförande studier mot referensläkemedlet krävs för att utesluta klinisk betydelsefulla skillnader.
Vilka faktorer påverkar omfattningen och typen av kliniska studier för godkännande av biosimilarer?
En faktor är molekylens komplexitet och tillgängliga jämförelsedata. För enklare molekyler med väl etablerad aktivitet (till exempel filgrastim) och när jämförande kvalitetsdata är robusta kan det vara tillräckligt att jämföra effekten av biosimilaren och referensläkemedlet genom studier av farmakokinetik och farmakodynamisk aktivitet hos friska frivilliga. För större molekyler som monoklonala antikroppar är en jämförande studie på patienter med ett konventionellt kliniskt effektmått vanligtvis nödvändigt. Vid tillgång till ett effektmått för farmakodynamisk aktivitet som uppvisar korrelation med effekten är konventionella effektmått i allmänhet inte nödvändiga om den farmakodynamiska aktiviteten korrelerar med läkemedlets effekt.
Vid säkerhetsproblem med referensläkemedlet eller inom den farmakologiska klassen så samlas säkerhetsdata under det kliniska utvecklingsprogrammet (även i samband med studier av farmakokinetik och farmakodynamisk aktivitet). Mängden data beror normalt på typen och allvarlighetsgraden hos de risker som identifierats för referensläkemedlet. Man utgår ifrån att farmakodynamiska biverkningar med biosimilaren förekommer med samma frekvens som med referensläkemedlet. För en biosimilar med potentiell immunogenicitet krävs analytiska studier, vanligtvis kompletterat med immunogenicitetsdata från kliniska studier. En faktor med biosimilarer som har diskuterats en hel del är möjligheten att extrapolera till andra indikationer. Detta innebär att existerande indikationer för referensläkemedlet kan godkännas för biosimilaren även om specifika kliniska studiedata för biosimilaren saknas. Detta kan accepteras om vetenskapliga belägg från jämförbarhetsstudier kan fastställa biosimilaritet och förutsatt att de specifika aspekterna (till exempel verkningsmekanism, potentiellt unika säkerhets eller immunogenicitetsaspekter) hos den ”extrapolerade” indikationen har beaktats. Extrapolering av data till andra indikationer ska alltid stödjas av robusta fysikalisk-kemiska studier och in vitro-studier för att utvärdera alla tänkbara verkningsmekanismer.
Extrapolering är inte ett nytt koncept, utan en väletablerad vetenskaplig princip som används rutinmässigt när biologiska läkemedel som är godkända för flera indikationer genomgår större förändringar i tillverkningsprocessen (till exempel introduktion av en ny sammansättning). I de flesta av dessa fall upprepas inte de kliniska prövningarna för alla indikationer, och förändringarna godkänns baserat på jämförande kvalitets- och in vitro-studier. Alla indikationer för biologiska läkemedel (inklusive biosimilarer) har beviljats på grundval av solida vetenskapliga belägg.
Vilka biosimilarer som används inom reumatologin är tillgängliga i Sverige år 2023?
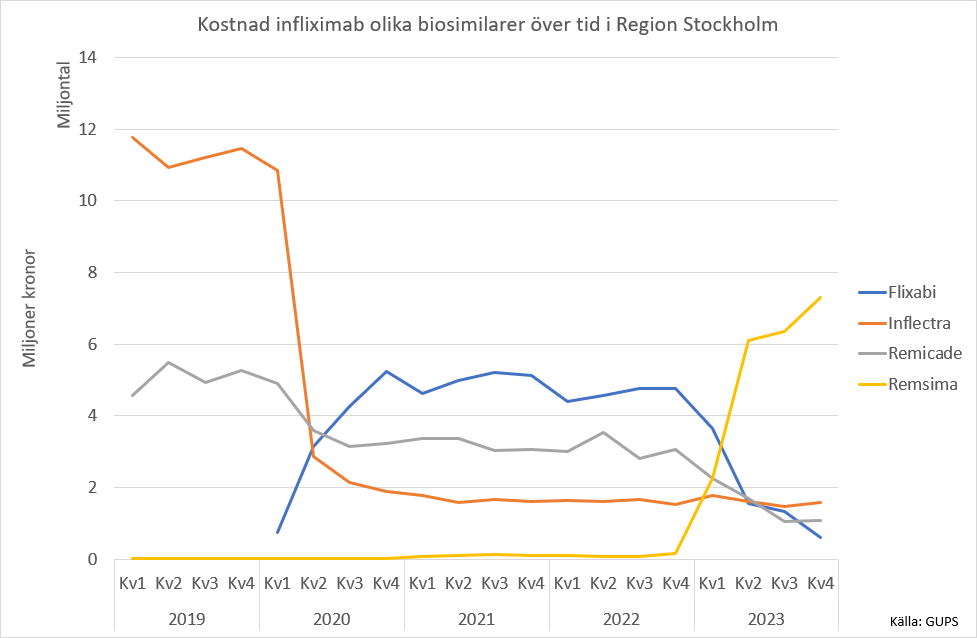
Infliximab
För infliximab är det tre olika biosimilarer CT-P13 (Inflectra eller Remsima), SB2 (Flixabi) och PF-06438179 (Zessly). När det gäller CT-P13 så marknadsförs samma substans från tillverkaren som befinner sig i Sydkorea av såväl Pfizer (Inflectra) som av Orion Pharma (Remsima). De kliniska studierna som låg till grund för godkännandet inkluderade en studie på patienter med reumatoid artrit (RA) [3, 4] och en annan på patienter med ankyloserande spondylit (AS) [5, 6]. I båda studierna randomiserades patienterna till CT-P13 eller Remicade under 30 veckor. Därefter erhöll alla patienter CT-P13 under ytterligare 48 veckor. Det visade sig att det inte förelåg några skillnader avseende ACR 20/50/70 respektive ASAS 20/40 mellan grupperna. Förekomsten av anti-drug-antibodies efter 30 veckors behandling i kombination med metotrexat av patienter med RA var lika hög med CT-P13 (48,4 procent) som med Remicade (48,2 procent).
Utifrån de regulatoriska kraven får man intrycket att de kliniska studierna var mera omfattande studier än nödvändigt. Anledningen till detta kan ha varit att man inte enbart ville övertyga läkemedelsmyndigheterna, men även förskrivarna som var ovana att använda biosimilarer.
De nordiska länderna förhöll sig något olika när den första biosimilaren för infliximab lanserades. Danmark var mycket proaktiv och ett läkemedelsråd för dyra sjukhusläkemedel (RADS) samordnade introduktionen av biosimilarer med registrering av batchnummer och rapportering av biverkningar samt uppföljning i nationella hälsodatabaser [7]. Inom loppet av ett år från början av 2015 till början av 2016 hade i princip all användning av Remicade flyttats över till biosimilaren CT-P13. Introduktionen av infliximab-biosimilaren i Sverige var avsevärt långsammare än i Danmark och ett år efter lanseringen av biosimilarer utgjordes endast 25 procent av infliximab av biosimilaren CT-P13. I Norge var andelen biosimilarer för infliximab cirka 60 procent, medan Finland knappt hade börjat använda biosimilarer. Två år efter att infliximab-biosimilarer blev tillgängliga låg andelen kring 65 procent i Sverige, nästan 100 procent i Norge och cirka 40 procent i Finland. En anledning till att bytet till biosimilarer var långsammare i Sverige var att man initialt valde att använda biosimilarer framför allt vid nyinsättning. En annan anledning var att man ville invänta resultaten från en 52 veckors norsk studie (NOR-SWITCH) [8] på patienter med olika indikationer för infliximab (RA, PsA, AS, SPA, ulcerös kolit, Mb Crohn, psoriasis). Patienter som stod på Remicade randomiserades till att fortsätta med Remicade eller byta (Switch) till biosimilaren CT-P13. Denna studie, som publicerades 2017 kunde inte påvisa några skillnader i effekt och även förekomsten av biverkningar var likartad i båda grupperna. Nästan samtidigt presenterades även danska registerdata (DANBIO) som visade att effekten av CT-P13 inte skiljer sig från Remicade [9].
Etanercept
När det gäller etanerceptbiosimilarerna så valde Sandoz, som tillhandahåller Erelzi (GP2015) att göra en klinisk studie på patienter med plack-psoriasis. Efter en tolv veckors behandlingsperiod randomiserades patienterna till Enbrel eller GP2015 under sex veckor, därefter erhöll alla patienter under sex veckor det preparat de först hade behandlats med och sedan fortsatte man under sammanlagt 52 veckor [10]. Man kunde inte se någon skillnad i effekt avseende PASI 50/75/90 mellan grupperna. Den andra etanercept-biosimilaren som marknadsförs av Biogen under namnet Benepali (SB4) studerades i en 52-veckorsstudie på patienter med RA, som under minst sex månader hade behandlats med metotrexat och prednisolon (<10 mg) [11, 12]. Effekten mellan preparaten var jämförbar. Även incidensen av biverkningar var jämförbar, men andelen patienter med erytem vid injektionsstället var något högre med Enbrel (11 procent) än med biosimilaren SB4 (2 procent).
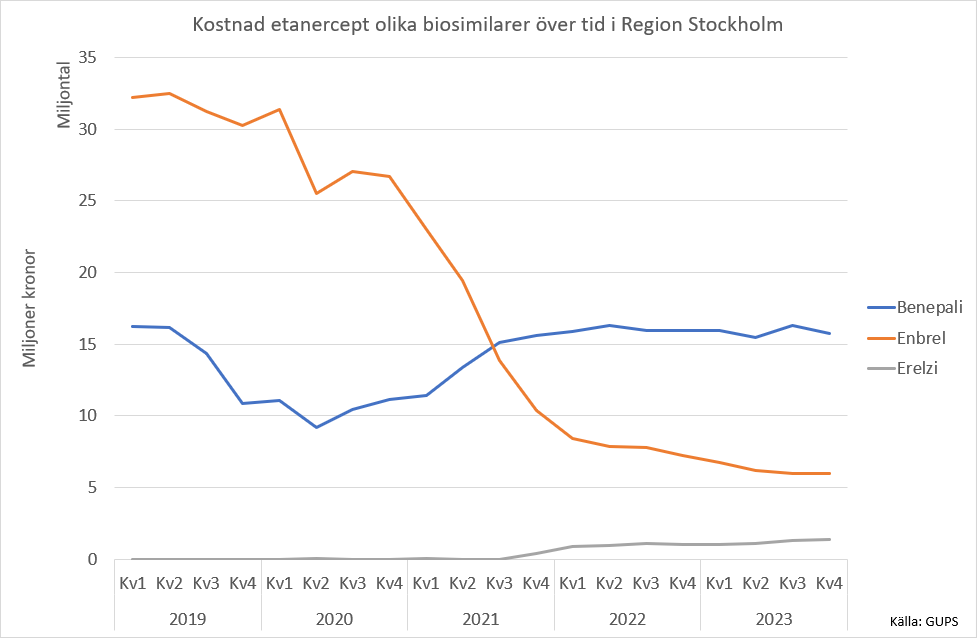
Introduktionen etanercept-biosimilarer var mycket snabbare i Danmark än i Sverige och Norge [7]. Inom loppet av ett halvår var andelen etanercept-biosimilarer 84 procent (initialt omfattades inte alla indikationer för Enbrel av etanercept-biosimilarer). I Sverige ökade användningen av etanercept-biosimilarer långsammare och 12 månader efter introduktionen var andelen cirka 60 procent. Och 24 månader efter introduktionen var andelen 50 procent.
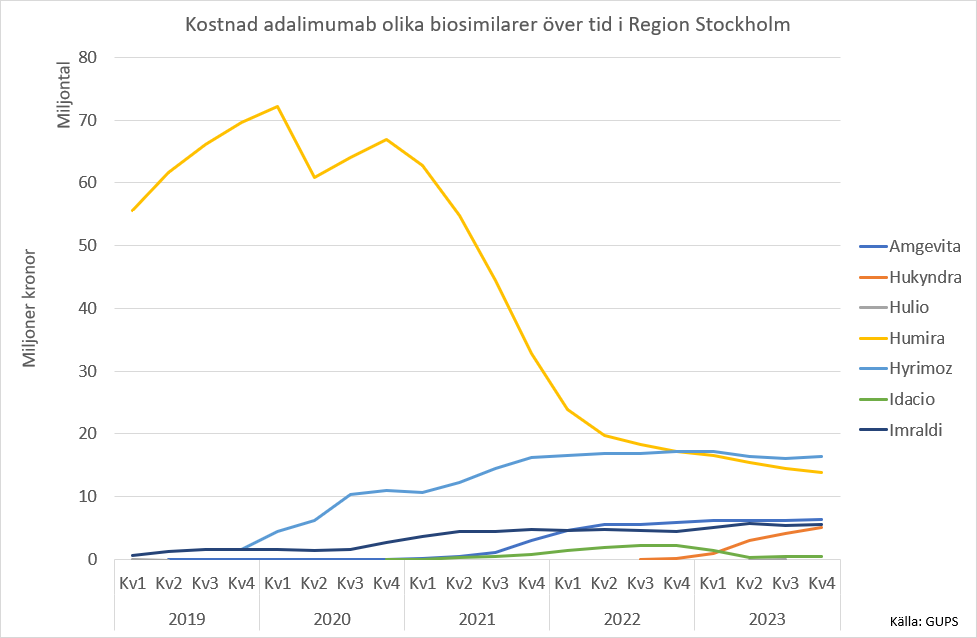
Adalimumab
Under 2018 introducerades biosimilarer för adalimumab och för närvarande finns sex biosimilarer för denna anti-TNF-antikropp (ABP501, AVT02, FKB327, GP2017, MSB11022 och SB5) [13]. I studierna har man valt lite olika design och för vissa preparat har man gjort studier med switch och utan switch och med olika indikationer. Men sammantaget har man inte kunnat visa att biosimilarerna skulle ha sämre effekt eller mera biverkningar än originalpreparatet. Och om man betraktar biosimilarerna som ytterligare en variant av ett redan befintligt läkemedel som inte skiljer sig åt mera än olika batcher av originalpreparatet är det kanske inte så förvånande. Ett flertal biosimilarer ökar konkurrensen och bidrar till att pressa priserna och man får hoppas att intresset för att utveckla biosimilarer håller i sig.
Region Stockholms läkemedelskommittés expertgrupp för smärta och reumatiska sjukdomar
Artikeln är tidigare publicerad i Reumabulletinen 28 februari 2024
Källa
- EMA. Biosimilarer i EU: Vägledning för vårdpersonal. 2020 [cited 2023-10-09]
- Läkemedelsverket. Analysera switch och i förlängningen utbytbarhet av biologiska läkemedel på apotek. 2023 [cited 023-10-09]
- Yoo DH, Prodanovic N, Jaworski J, Miranda P, Ramiterre E, Lanzon A, et al. Efficacy and safety of CT-P13 (biosimilar infliximab) in patients with rheumatoid arthritis: comparison between switching from reference infliximab to CT-P13 and continuing CT-P13 in the PLANETRA extension study. Ann Rheum Dis. 2017 Feb;76(2):355-363
- Yoo DH, Racewicz A, Brzezicki J, Yatsyshyn R, Arteaga ET, Baranauskaite A, et al. A phase III randomized study to evaluate the efficacy and safety of CT-P13 compared with reference infliximab in patients with active rheumatoid arthritis: 54-week results from the PLANETRA study. Arthritis Res Ther. 2016 Apr 2;18:82
- Park W, Yoo DH, Jaworski J, Brzezicki J, Gnylorybov A, Kadinov V, et al. Comparable long-term efficacy, as assessed by patient-reported outcomes, safety and pharmacokinetics, of CT-P13 and reference infliximab in patients with ankylosing spondylitis: 54-week results from the randomized, parallel-group PLANETAS study. Arthritis Res Ther. 2016 Jan 20;18:25
- Park W, Yoo DH, Miranda P, Brzosko M, Wiland P, Gutierrez-Ureña S, et al. Efficacy and safety of switching from reference infliximab to CT-P13 compared with maintenance of CT-P13 in ankylosing spondylitis: 102-week data from the PLANETAS extension study. Ann Rheum Dis. 2017 Feb;76(2):346-354
- Jensen TB, Bartels D, Sædder EA, Poulsen BK, Andersen SE, Christensen MMH, et al. The Danish model for the quick and safe implementation of infliximab and etanercept biosimilars. Eur J Clin Pharmacol. 2020 Jan;76(1):35-40
- Jørgensen KK, Olsen IC, Goll GL, Lorentzen M, Bolstad N, Haavardsholm EA, et al. Switching from originator infliximab to biosimilar CT-P13 compared with maintained treatment with originator infliximab (NOR-SWITCH): a 52-week, randomised, double-blind, non-inferiority trial. Lancet. 2017 Jun 10;389(10086):2304-2316
- Glintborg B, Sørensen IJ, Loft AG, Lindegaard H, Linauskas A, Hendricks O, et al. A nationwide non-medical switch from originator infliximab to biosimilar CT-P13 in 802 patients with inflammatory arthritis: 1-year clinical outcomes from the DANBIO registry. Ann Rheum Dis. 2017 Aug;76(8):1426-1431
- Gerdes S, Thaçi D, Griffiths CEM, Arenberger P, Poetzl J, Wuerth G, et al. Multiple switches between GP2015, an etanercept biosimilar, with originator product do not impact efficacy, safety and immunogenicity in patients with chronic plaque-type psoriasis: 30-week results from the phase 3, confirmatory EGALITY study. J Eur Acad Dermatol Venereol. 2018 Mar;32(3):420-427
- Emery P, Vencovský J, Sylwestrzak A, Leszczyński P, Porawska W, Baranauskaite A, et al. A phase III randomised, double-blind, parallel-group study comparing SB4 with etanercept reference product in patients with active rheumatoid arthritis despite methotrexate therapy. Ann Rheum Dis. 2017 Jan;76(1):51-57
- Emery P, Vencovský J, Sylwestrzak A, Leszczynski P, Porawska W, Baranauskaite A, et al. 52-week results of the phase 3 randomized study comparing SB4 with reference etanercept in patients with active rheumatoid arthritis. Rheumatology (Oxford). 2017 Dec 1;56(12):2093-2101
- Abitbol V, Benkhalifa S, Habauzit C, Marotte H. Navigating adalimumab biosimilars: an expert opinion. J Comp Eff Res. 2023 Nov;12(11):e230117
Senast ändrad